
Fenylketonurie – co je to onemocnění, proč se vyskytuje a jak zacházet se strouhou?
![]()

Poté, co jsme zjistili, jaký druh onemocnění – fenylketonurie, diagnostikovaný během novorozeneckého období, je nutné okamžitě zahájit léčbu, když je zjištěna. Včasná detekce a terapie umožňují dosažení příznivých výsledků.
Fenylketonurie – co je tohle onemocnění?
Fenylketonurie, nebo těžebních nemoc, je závažné patologie, poprvé popsána v roce 1934 norský vědec Felling. Potom se provede průzkum Fellinge málo dětí s mentální retardací a identifikovat jejich přítomnost v fenylpyruvátu moči – produkt rozkladu aminokyseliny fenylalaninu dodávané s potravinami, který je v těle pacientů není rozdělen. Fenylketonurie – onemocnění spojená s metabolickými poruchami přirozené povahy, otevřený jeden z prvních.
Fenylketonurie je druh dědičnosti
Fellingova choroba je chromozomálně-genetická, dědičná, přenášená dětem od rodičů. Viníkem pro vývoj patologie je gen nacházející se na 12 chromozomu. Je zodpovědný za produkci jaterního enzymu fenylalanin-4-hydroxylázy, kterým přenese fenylalanin na jinou látku – tyrosin (vyžaduje se pro normální fungování těla).
Bylo zjištěno, že fenylketonurie je dědičná jako recesivní charakter. Přibližně 2% lidí, kteří jsou nositeli vadného genu, ale netrpí fenylketonurií. Patologie vyvíjí pouze tehdy, pokud matka a otec dítěte předat gen, ale to se může stát s pravděpodobností 25%. Pokud PKU se dědí jako recesivní, heterozygotní ženou a manžel je homozygotní pro normální alelu genu, pak je pravděpodobné, že děti budou zdravými nositeli fenylketonurie genu je 50%.
Formy fenylketonurie
Vzhledem k tomu, kdo může vyvinout fenylketonurie, jaký druh nemoci, často je to klasická forma patologie, která se vyskytuje v přibližně 98% případů. Zbývající případy – kofaktorová fenylketonurie způsobená poruchou tetrahydrobiopterinu v důsledku porušení jeho syntézy nebo obnovení aktivní formy. Tato látka slouží jako kofaktor řady enzymů a bez ní není možné projevit jejich činnost.

Fenylketonurie – příčiny
Kácení onemocnění je patologie, ve které v důsledku mutací v genu způsobuje nedostatek nebo nepřítomnost fenylalanin 4-hydroxylázy, se hromadí v tkáních a tělních tekutinách fenylalaninu a jeho dílčích produktů štěpení. Část přebytku fenylalaninu se převede na keton, fenylovou výstupu moči, která určila název nemoci.
Porucha metabolických procesů postihuje hlavně mozek. Na jeho tkáních vzniká toxický účinek, procesy metabolismu tuků jsou narušeny, myelinizace nervových vláken selhává a tvorba neurotransmiterů se snižuje. Začíná tak spuštění patogenetických mechanismů mentální retardace u dítěte.
Fenylketonurie – příznaky
Při narození vypadá dítě s touto diagnózou zdravé a až po 2-6 měsících se objeví první příznaky. Fenylketonurií symptomy se začne zobrazovat, když je v těle dítěte hromadí fenylalanin, přichází spolu s mateřským mlékem nebo vzorec pro umělou výživu. Mohou existovat takové, které ještě nejsou konkrétní příznaky:
- nadměrná letargie nebo úzkost;
- bezvýznamné pláče;
- svalová dystonie;
- častá regurgitace;
- křeče;
- poruchy spánku.
Navíc nemocné děti mají lehčí pokožku, vlasy a oči než zdravé rodinné příslušníky, což je spojeno s porušením výroby pigmentového melaninu v těle. Dalším diagnostickým příznakem, který lékaři nebo pozorní rodiče mohou zaznamenat, je druh “myší” pachu způsobený uvolněním fenylalaninu v moči a potu.
Klinické projevy se projevují více než šest měsíců po zavedení prvního doplňkového jídla:
- neschopnost zaměřit se na jednotlivé předměty;
- lhostejnost vůči všemu, co se děje;
- absence výrazů obličeje, úsměvy;
- třes rukou a tak dále.
Pozoruhodné jsou také fyzické abnormality: malá velikost hlavy, vynikající horní čelist, zpomalení růstu. Nemocné děti později začnou držet hlavu, plazit, posadit se a vstát. Zvláštní postavení v sedící pozici je typické – “krejčí” postoj, s pažími neustále ohnutými na loktech a nohama na kolenou. Ve věku tří let, pokud nebyla léčba zahájena, se symptomatologie zvyšuje.

Fenylketonurie – diagnostika
Fenylketonurie u dětí je často zjištěna v mateřské nemocnici, což umožňuje zahájit léčbu včas a zabránit vzniku řady nezvratných následků. Po 4-5 dnech po porodu se děti na kapkují krve na prázdný žaludek, aby zjistily některé závažné genetické nemoci, mezi nimi i fenylketonurie. Pokud se výpis z mateřského nemocnice objevil dříve, analýza se provádí v polyklinice v místě bydliště během prvních deseti dnů života.
Vzhledem k tomu, že v ojedinělých případech existují chybné výsledky, diagnóza se po výsledcích první analýzy nikdy nestanoví. Pro potvrzení stávající patologie je přiděleno několik dalších studií, mezi které patří:
- analýza moči pro detekci fenylpyruvátu;
- kvantitativní stanovení fenylalaninu a tyrosinu v plazmě;
- stanovení aktivity jaterních enzymů;
- elektroencefalografie a zobrazování mozku magnetickou rezonancí.
Genetický defekt vedoucí k vývoji patologie může být zjištěn u plodu během invazivní prenatální diagnostiky. K tomu jsou vybrány vzorky buněk z chorionu villus nebo amniotickej tekutiny a poté je provedena analýza DNA. Doporučuje se taková studie v rodinách s vysokým rizikem nemocnosti, včetně případů, kdy již existuje dítě s fenylketonurií.
Fenylketonurie – léčba
Když je u novorozenců zjištěna fenylketonurie, lékaři takových specialit, jako je genetik, pediatr, neurolog, výživový lékař, by měli pozorovat nemocné. Ti, kdo znají, fenylketonurie – jaký druh onemocnění, bude jasné, proč je základem jeho léčby dodržovat dietu s omezením fenylalaninu. Kromě toho jsou předepsány léky, masáže, fyzioterapie, psychologické a pedagogické metody pro socializaci dítěte, příprava na učení.

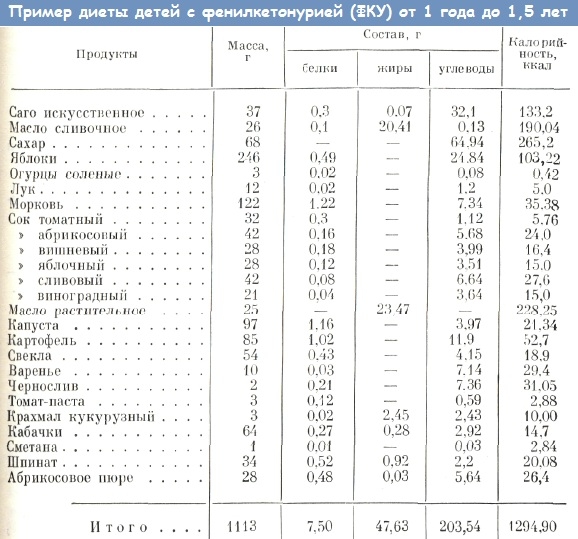
Fenylketonurie – dieta
Při diagnostikování “fenylketonurie” je okamžitě předepsána výživa pro dítě. Vyloučeny z dietních potravin bohatých na bílkoviny (maso, ryby, mléčné výrobky, luštěniny, ořechy, atd.) Proteinové potřeby kompenzovány dietní směsi a jiné výrobky s berlofenom – polosyntetický proteinový hydrolyzát, zcela bez fenylalaninu (Tetrafen, Lofenalak, Nofelan). Pacienti berou bez bílkovin chléb, těstoviny, obiloviny, pěny a tak dále. Kojení se provádí v omezených dávkách.
Přísné dodržování stravy s pravidelným sledováním obsahu fenylalaninu v krvi během prvních 14 až 15 let života zabraňuje rozvoj mentálních abnormalit. Poté je dieta poněkud rozšířena, ale mnozí odborníci doporučují celoživotní dodržování zvláštní stravy. Kofaktorová forma fenylketonurie není léčena dietou, ale je korigována pouze podáváním přípravků tetrahydrobiopterinu.
Fenylketonurie – léčivé přípravky
Léčba fenylketonurií u dětí také zajišťuje příjem některých léků, včetně:
- nootropika (Piracetam, Cerebrolysin);
- vitaminy skupiny B;
- minerální komplexy;
- léky pro zlepšení tkáňového metabolismu (ATP, Riboxin);
- léky, které zlepšují mikrocirkulaci (Trental, Pentoxifylline).
Fenylketonurie – prognóza života a onemocnění
Rodiče, kteří znají z první ruky, jaké genetické onemocnění – fenylketonurie, jsou v moderních podmínkách příležitostí k růstu zdravého dítěte, pokud budete dodržovat všechny lékařské předpisy. Pokud není k dispozici správná léčba, prognóza fenylketonurie je zklamáním: pacienti žijí přibližně 30 let se závažnou mentální nedostatečností a více funkčními poruchami.